
Targeting cortical circuit dysfunction to restore movement in Huntington's disease mice
17 March 2026
Huntington’s disease is a devastating movement disorder without a cure. Although the genetic cause is well-defined, the complex downstream effects that underlie the symptoms are poorly understood.
Dr Sonja Blumenstock from the University of California San Diego, recently spoke about her work at SWC as a winner of the Emerging Neuroscientists Seminar Series. She investigates cortical dysfunctions in Huntington’s disease mice, focusing on how different neuron subtypes are affected as the disease progresses, and how this shapes the animal’s movements.
Using two-photon calcium imaging, she examined the activity of two cortical inhibitory neuron subtypes and excitatory corticostriatal projection neurons in the motor cortex. Motor deficits were accompanied by reduced activity in one of the inhibitory subtypes and in the downstream corticostriatal neurons. Targeted activation of inhibitory neurons rescued these deficits, pointing to a potential therapeutic target in Huntington's disease. In this Q&A, she discusses her findings.
You’ve found that activity in a class of interneurons is reduced in Huntington's disease mice, and that tracks with a decline in motor skills. Can you tell us more – was it a surprise to see that?
Yes, I was surprised to see that the activity of vasoactive intestinal peptide (VIP) interneurons was reduced; they're hypoactive during movement. It came as a surprise in two ways.
First of all, they're not generally thought to play a major role in Huntington’s disease. They weren't considered particularly vulnerable. They don't die early, and they don't seem to be strongly affected.
The second surprise was the magnitude of change in their activity. It was the opposite of what they should normally be doing during motor behaviour.
I did think it was strange, and I wondered if I was doing something wrong. But after checking my analysis and independently reproducing the result several times, I realised that this was an important finding.
You then moved to stimulating these cells – why these inhibitory cells and not the downstream neurons or others within the same circuit?
All my disease research builds on a large body of knowledge from basic research. I knew that VIP cells are important for learning. It is known that they become active when an animal is doing something new, when they're attending to something, when they're aroused and when they want to move.
So when I activated them, that was the goal that I had in mind. I want to facilitate a pathway that is known to be important in learning, without relying on repairing a cell type that is severely degenerated, but instead boosting a pathway that is still present but functionally impaired. That’s something you want in a Huntington's disease scenario.
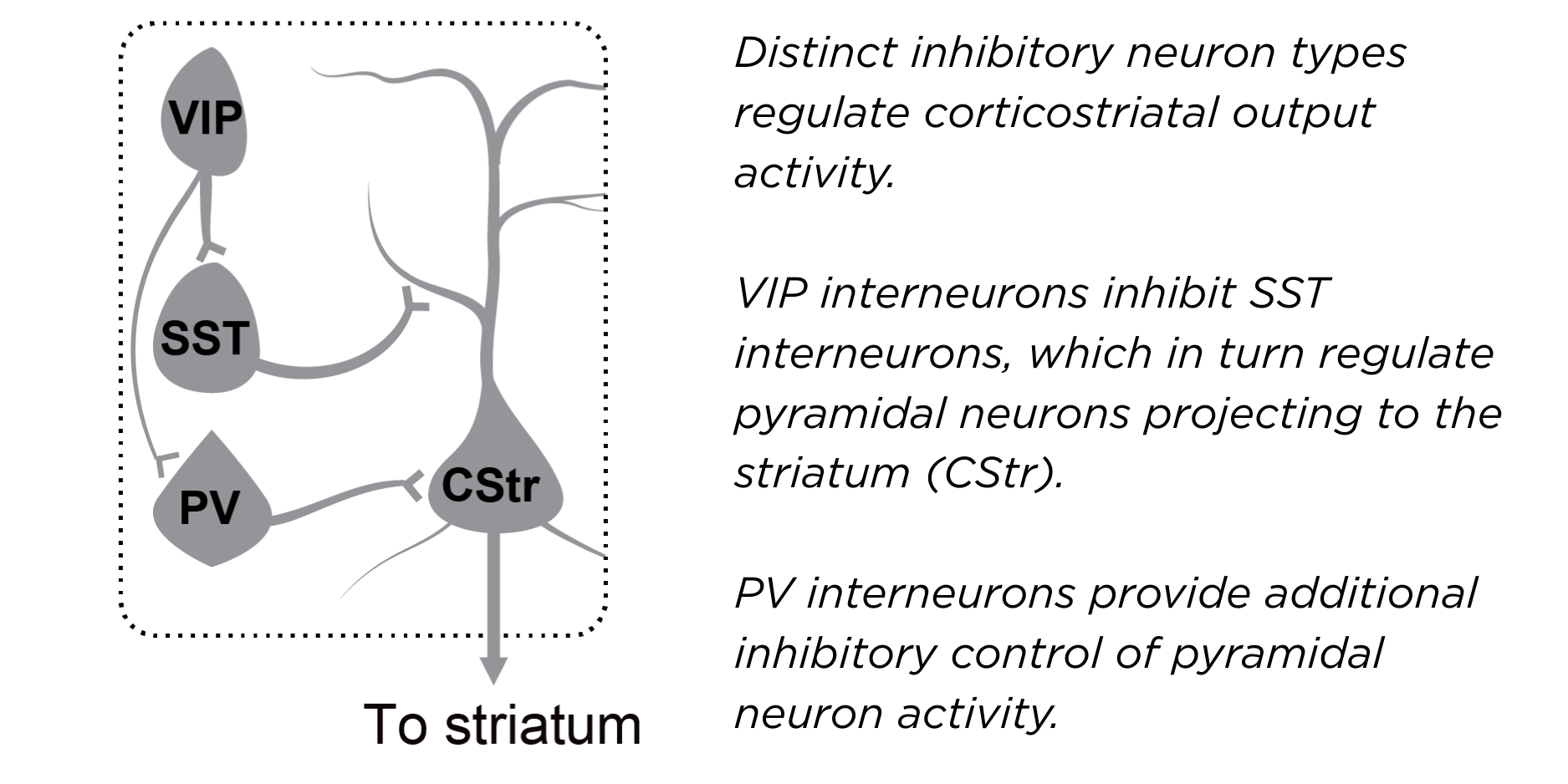
Diagram showing how inhibitory neuron types regulate corticostriatal output activity.
You showed that activating those cells led to sustained improvements in motor performance. What did that look like?
Using optogenetics, I stimulated these cells every second day for only a few minutes per session. This was during a task where the mice had to repeatedly move in a controlled way.
I did that for a total of three weeks. Over that time, gradually, the mice would start learning the task better than the mice that I didn't stimulate. The beneficial effects lasted as long as I looked.
It's important to point out that this circuit repair doesn’t cure Huntington’s disease. This is one piece of the puzzle to treat a very complex disease – by itself, it doesn’t stop the progression of the disease.
You're working with mice. How confident are you that the same VIP interneuron dysfunction is happening in Huntington’s in humans?
I am confident in the sense that one upstream input into VIP cells – cholinergic modulation – is reduced in humans in Huntington’s disease. Because VIP interneurons are strongly driven by cholinergic inputs, I would expect this to affect their function. However, it is not currently possible to measure VIP activity directly in the human cortex.
There are also several studies showing interneuron degeneration in human cortex in Huntington’s disease. Some reports suggest a stronger loss of somatostatin interneurons than the VIP cells, which is notable because VIP interneurons preferentially connect to somatostatin cells. So at this point, most of the human evidence is pathological or molecular rather than functional, but there is indirect evidence pointing to disruption of this circuit.
Can you speculate on what the path might look like to target VIP interneurons with therapies in humans? Could existing tools like deep-brain stimulation or targeted therapies approximate what you've achieved in mice?
Deep brain stimulation is being explored for Huntington's disease, but not in the cortex. More generally, many neurological therapies already work by modulating specific neuromodulatory receptors rather than directly stimulating neurons. If we think about how to increase the activity of VIP interneurons specifically, a more promising path might be pharmacological approaches that target receptors enriched in these cells.
For example, VIP interneurons express the gastrin-releasing peptide receptor, which can be activated pharmacologically in mice. They also express subsets of cholinergic receptors, which could potentially be modulated using positive allosteric modulators. These types of drugs would enhance the remaining context-dependent inputs that VIP cells normally receive, even if those signals are weaker in Huntington’s disease. That's something that I would like to explore in the future, because it could potentially approximate the circuit activation we achieved with optogenetic stimulation in mice.
Fluorescent labelling of different neuronal populations, illustrating the diversity of cell types that contribute to cortical circuit function.
In your talk, you focused on motor circuits, but also mentioned that you've seen the VIP neurons dysfunctional in other areas of the brain, too. Could you expand on that, and what else is happening in the brain in Huntington's disease?
I did see reduced VIP activation in other areas of the cortex, like the visual cortex, and somatosensory cortex.
VIP cells are known to be globally active across the cortex during movement. It has been studied particularly well in the visual cortex, even though their activity there is strongly related to movement.
The most widely studied circuit dysfunctions in Huntington's disease are within the striatum because that region is affected most severely. There is an imbalance between two different cell types – direct and indirect pathway spiny neurons – which send different outputs through the basal ganglia to facilitate movement or inhibit unwanted movement. These two cell types normally maintain that balance and are very important for movement control.
One of these populations is more vulnerable in Huntington's disease and degenerates first, which throws the entire circuit out of balance. In addition, the connections from the cortex to the striatum also degenerate early. This may further drive the circuit imbalance.
There are many other factors contributing to dysfunction, such as the reduction in BDNF, brain-derived neurotrophic factor, which is important for brain health. Like other neurodegenerative diseases, Huntington's disease is multifactorial.
Do your findings about the role of VIP interneurons add to how we might think about other neurodegenerative conditions?
I have started to look at healthy ageing, which can be associated with a reduced ability to learn. We can even consider VIP cells as a target for modulation there. But for the individual diseases, I think the first step is always to determine the nature of the dysfunction.
What are you planning to focus on next?
With this VIP stimulation, we seem to increase plasticity in the brain. And through this plasticity mechanism, behaviour improves and circuit function improves. That's something I want to show experimentally - does plasticity actually get better? To address this, I will pair the VIP stimulation with synaptic analysis. That’s the immediate focus.
About Dr Blumenstock
Sonja completed her undergraduate training in Biochemistry and earned a Master’s degree in Experimental and Clinical Neuroscience at the University of Regensburg, Germany. She received her PhD in 2017 from the German Center for Neurodegenerative Diseases (DZNE), where she investigated mechanisms of synaptic decline in neurodegeneration. She subsequently joined the laboratory of Irina Dudanova at the Max Planck Institute of Biological Intelligence as a postdoctoral researcher, focusing on molecular and circuit signatures of Huntington’s disease. In 2021, she established a research collaboration with the laboratory of Takaki Komiyama at the University of California San Diego as a Visiting Scholar and formally joined the lab in 2024 as an Assistant Project Scientist. Her research integrates longitudinal in vivo imaging, optogenetics, and behaviour to uncover cell-type-specific circuit mechanisms underlying motor control and neurodegenerative disease.


